
1Эрдэм Шинжилгээний Төв Лаборатори, Анагаах Ухааны Хүрээлэн, Монгол улс, 2Мэдрэлийн Эмгэг Судлалын Салбар, Анагаах Ухааны Хүрээлэн, Монгол улс, 3Анагаах Ухааны Салбарын Амьдралын Молекулын Үндсийн Ухааны Тэнхим, Токай Их Сургууль, Япон улс, 4 Мэдрэлийн Эмгэг Судлалын Тэнхим, Токушимагийн Их Сургууль, Япон улс
An interesting case of a hereditary neuromuscular disease have been revealed in Dornod province. A total of 28 individuals in 3 families have showed the symptoms of hereditary neuromuscular disease. The final diagnosis couldn't be established due to similarities of symptoms between Friedreichs' ataxia and Charcot-Marie-Tooth disease. Friedreichs' ataxia is a relatively rare disease with a distribution of I'OOO'OOO : 1 in south-east Asia, whereas Charcot-Marie-Tooth disease, motor and sensory neuropathy characterized by distal muscle weakness and atrophy, occurs in 2'500 : 1.
A total of 114 individuals from 3 families have participated in our study. 21 affected and 15 unaffected family members were selected for advanced molecular genetic study. The selection of individuals from total people were based on the results of genetic pedigree, neurological examination and neurophysiologic analysis.
10 ml of blood sample was obtained from peripheral vein using the Vacutainer system. Using the Genome QIAmp DNA blood kit (QIAGEN) the genomic DNA was extracted from the blood sample, the degradation of which was checked by the gel electrophoresis. The optical density measurement was performed for protein contamination and the concentration of the genomic DNA was measured by PicoGreen fluorescence assay. Using the quantitative PCR on ABI 7000 we have tested the duplication of Peripheral Myelin Protein 22 (PMP22) gene, located on 17p11 locus. Also we have designed the TaqMan probe for ехопЗ of PMP22 gene and an internal control - a TaqMan probe for ехопЗ of gene coding the serum Albumin protein.
The result of the quantitative PCR have revealed the copy numbers of PMP22 gene among all patients were 1.5 folds of normal and the deletion of HNPP gene was determined. Basing on the family tree of these 3 families, we estimated that all these patients are heterozygote.
We concluded that the patients in this family have the duplication of PMP22 gene and the deletion of HNPP gene. These mutations are major etiologic factors of Charcot-Marie-Tooth disease type 1A (CMT1A). The diagnosis of Freidriechs' ataxia was denied, which reconfirms the results of other researchers, stating the rare occurrence of this disease in south east Asia.
Pp. 7-10, Table 1, Picture 1, Figures 2, References 10.
Удиртгал. Дорнод аймагт удамшлын мэдрэл булчингийн эмгэгтэй 28 хүн 3 гэр бүлд илэрсэн юм. Энэхүү тохиолдол нь Шарко-Мари-Тут зэрэг бусад удамшлын мэдрэл булчингийн эмгэгүүдтэй эмнэлзүйн шинж тэмдэг ижил тестэй байсан. (1) Шарко-Мари-Тут (ШМТ) нь арагийн дисталь хэсгийн булчинг хатингаршуулж тулгуур эрхтэн системийн Тажиг үүсгэх ба шөрмөсний гүний рефлекс үгүй болж хөделгөений болон мэдрэхүйн өөрчлөлтөнд оруулдаг эмгэг юм. ШМТ өвчний тархалтын давтамжийн талаарх мэдээллүүдээс үзэхэд нилээд ялгаатай байдаг. Удамшлын мэдрэл булчингийн эмгэгийн бүтэц, тархалтын давтамж Монгол орны бүс нутгуудад ч харилцан адилгүй байгааг судлаачид тэмдэглэсээр байна. (2) Е.Д. Маркова, Р.В. Магжанов, 1990, нарын хэвлэлийн тоймд ШМТ-н тархалт 100.000 хүн амд 2-5 давтамжтай боловч зарим тусгаарлагдмал (изолят) хүн амын дунд 1/50.000-аас 1/20.000 (Баруун Норвеги, Гуамын болон Хойд Еленагийн арлуудад) давтамжтай тохиолддог. Skre нар, 1974, ШМТ өвчний тархалтыг 2500 хүн амд 1 тохиолдлын давтамжтай гэж мэдээлсэн бол Emery нарынхаар, 1991, 10.000 хүн амд 1 тохиолдлын давтамжтай тохиолддог байна. ШМТ-н I ба II хэвшинжүүд нь 2:1 харьцаатайгаар I хэв шинж нь давамгайлж байдаг.
Ш. Батчулуун, 1990, Монгол орны хүүхэд насны хүн амын дунд мэдрэл булчингийн өвчний тархалтыг судалсан дүнгээс үзэхэд ШМТ нь 100.000 хүн амд 2 тохиолдлын давтамжтай байна (7).
Удамшлын мэдрэл булчингийн өөр нэгэн эмгэг болох Фридрихийн атаксийн үед төмөр-хүхэр хамааралт ферментийн тогтолцоо, эсийн амьсгал, исэлдэлтийн процесст • оролцдог фратаксин уургын нийлэгжилт алдагддаг. Фратаксин уургыг нөхцөлдүүлэгч ген, геномын хэсэг болох ГАА гурвал хэвийн хэмжээнээс хэд дахин илүүтэйгээр олшролт болсон байна. Эмнэлзүйн шинж тэмдэгийн хувьд ШМТ-тай төстэй, арагийн булчинг хатингаршуулж тулгуур эрхтэн системийн гажиг үүсгэхээс гадна зүрх, элэг, бөөр, нойр булчирхай зэрэг цуллаг эрхтэний үйл ажиллагааг алдагдуулдаг байна. Хамгийн түгээмэл тохиолдох эмнэлзүйн шинжтэмдэг нь нугас, тархины гаралтай тэнцвэргүйдэл юм. Romeo нар, 1983, Италид Фридрихийн атакси 22'000:1-25'000:1 тархалтын давтамжтай тохиолдож байна. Koenig нар, 1988, Кавказ үндэстэнд 50'000:1, африкийн орнуудад ховор тохиолддог. Dean нар, 1988, Кипрт их давтамжтай тохиолдож байна. Hirayama нар, 1994, Фридрихийн атаксиийн тархалтын давтамж нь 100'000:2-10 тохиолддог бөгөөд харин Зүүн Азийн улс орнуудад бараг тохиолддоггүй (100'000:0.1) гэж тэмдэглэжээ (8).
Зүүн Азийн улс орнуудад маш ховор тохиолддог эмгэг болох Фридрихийн атакси өвчний эмнэлзүйн хам шинж Дорнод аймгийн 3 гэр бүлийн нийт 28 хүнд илэрсэн нь бидний анхаарлыг зүй ёсоор татлаа.
Судалгааны материал. Дорнод аймгийн удамшлын мэдрэл булчингийн эмгэг бүхий 3 гэр бүлийн нийт 114 хүнийг судалгаанд хамруулсан. (Удмын зураглал) Удамзүйн зураглал, эмнэлзүйн нарийн мэргэжилийн эмчийн үзлэг, шинжилгээг үндэслэнэмгэгөөрчлөлтилэрсэнбуюутохиолдолын бүлэгт 21, эрүүл буюу хяналтын бүлэгт 15 хүнийг сонгож удамзүйн шинжилгээнд нийт 36 сорьц цуглуулав. Судалгааг Токай Их Сургууль, Анагаах Ухааны Хүрээлэн, Эрүүл Мэндийн Яамны ёс зүйн хороогоор тус тус оруулан баталсан болно. Судалгаанд оролцогч бүрт хийгдэх судалгааны талаар таниулж тайлбарласны дараагаар сайн дурын үндсэн дээр таниулсан зөвшөөрлийг авсан.
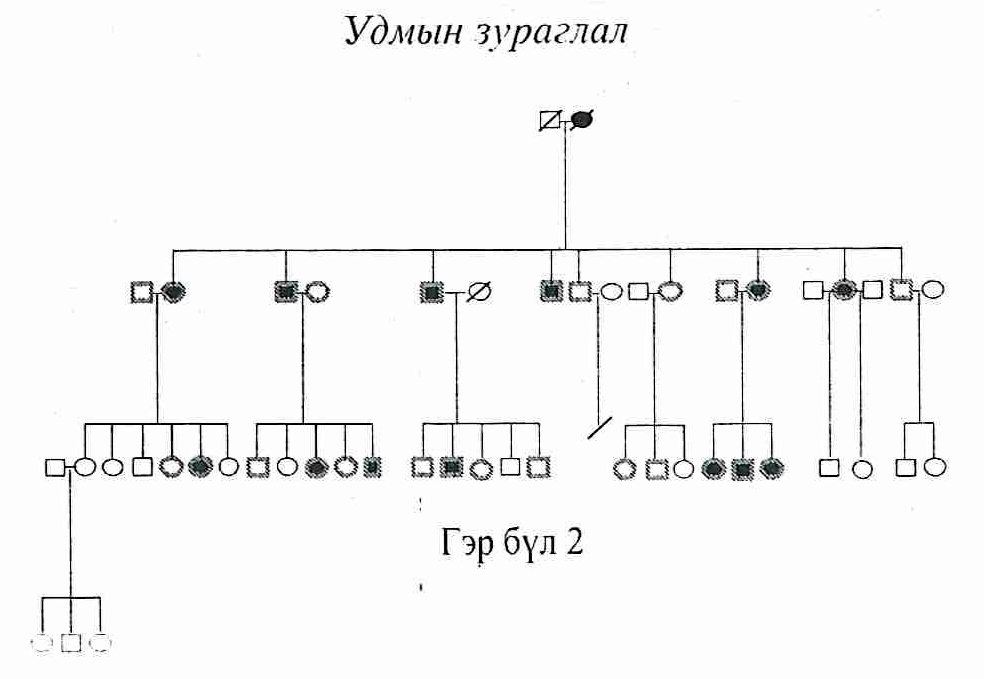
Судалгааны арга аргачлал. Асуумжийн аргаар удмын зураглалыг нарийвчлан зурсан. Эмнэлзүйн нарийн мэргэжилийн эмчийн үзлэг, булчингийн сэрэл дамжуулалтыг тодорхойлох электромиографийн аппаратын шинжилгээг Дорнод аймгийн Нэгдсэн Эмнэлгийн Мэдрэлийн
Тасаг, Анагаах Ухааны Хүрээлэнгийн Мэдрэл Судлалын Сектор, Токай Их Сургуулийн Мэдрэлийн Тэнхимийнэмчнарынбагхамтранхийжгүйцэтгэсэн. Судалгаанд хамрагдагсадаас удамзүйн зураглал, эмнэлзүйн нарийн мэргэжилийн эмчийн үзлэг, шинжилгээг ундэслэн эмгэг өөрчлөлт илэрсэн буюу тохиолдолын бүлэгт 21, эрүүл буюу хяналтын бүлэгт 15 хүнийг сонгож молекул генетикийн шинжилгээний сорьцыг цуглуулсан. Судалгаанд оролцогчийн захын цуснаас вакутейнер систем ашиглан 10мл-ыг сорьц болгон авсан. Сорьцноос геномын ДНХ-г QIAamp Maxi kit, QIAGEN ашиглан ялгасан. Геномын ДНХ-ийн цэвэршилт гарцыг спектрофотометр болон пикогрины аргаар тодорхойлж гель электрофорезоор шалгасан. Нөхцөлдүүлэгч ген, геномын хэсгийг TagMan сорилыг ашиглан тодорхойлсон. TagMan сорилын дотоод хяналт болгож альбумины генийг ашигласан. Статистик боловсруулалтыг стандарт хазайлтын дундаж утгуудыг тооцож хийсэн.
Хүснэгт 1
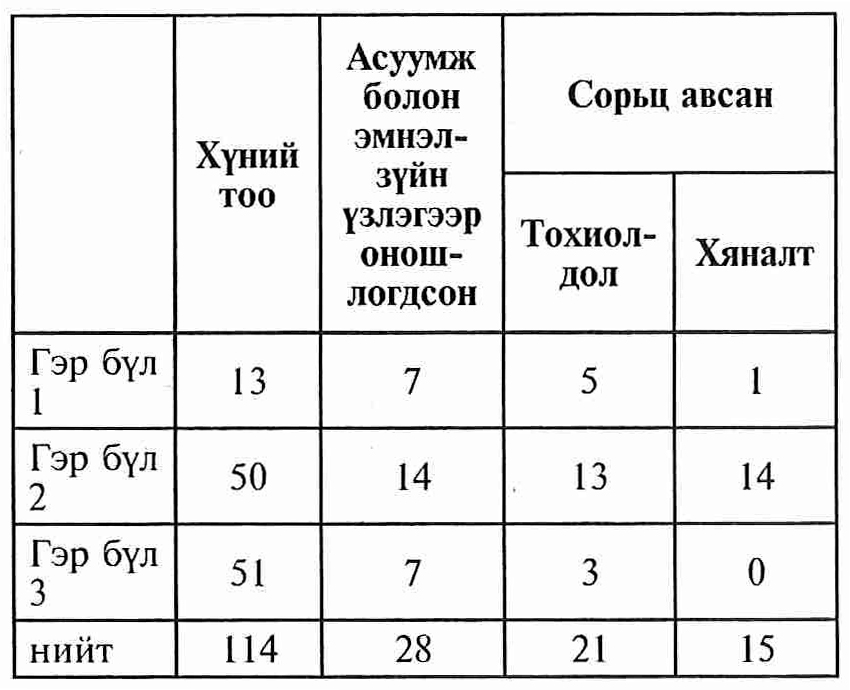
Үр дүн
- Удмын зураглалаар аутосомын доминант хэлбэрээр удамшсан байсан. (Удмын зураглал)
- Эмнэлзүйн нарийн мэргэжилийн эмчийн үзлэг шинжилгээгээр 3 гэр бүлийн гишүүдээс 28 хүнд дараах өвөрмөц шинж тэмдэгүүд илэрсэн:
- Шөрмесний рефлексүүд суларсан буюу арилсан шинж
- Фридрих төст тавхай
- Булчингийн хатингаршил
- Кифосколиоз (Зураг-1)
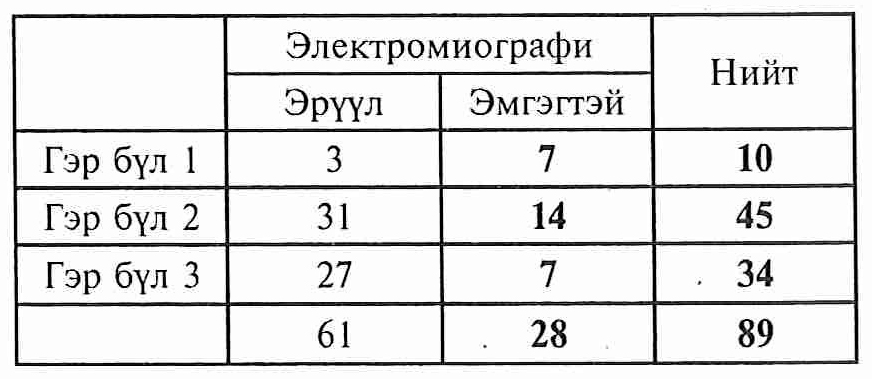
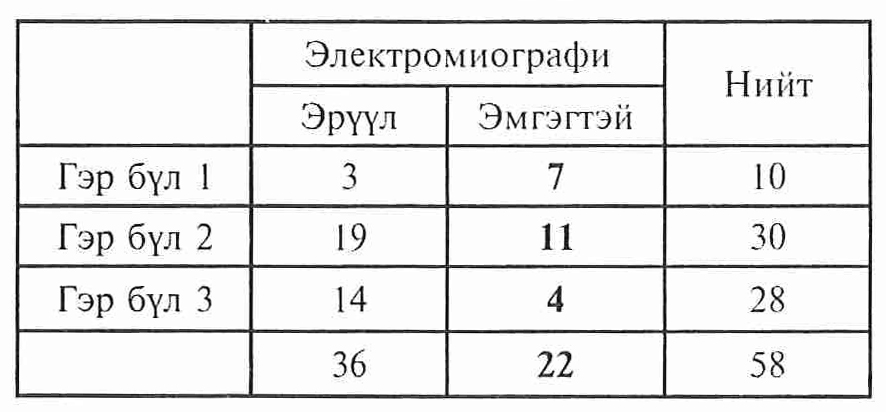
ЭМГ-ийн шинжилгээний үр дүн 3 гэр бүлийн гишүүдээс 22 хүнд ШМТ төст шинжүүд гарсан.
- Молекул генетикийн шинжилгээгээр нөхцөлдүүлэгч ген, геномын хэсгийг тодорхойлоход 17-рхромосомын богиномөрний 11-р локусд байрлах Захын Миелин Ширхэгийн Уургыг (Peripheral Myelin Protein 22, РМР22) кодлогч 1500 хос суурын урттай дараалал нь хоёрчлогдсон; удамшлын шалтгаантай Саа саажилтыг кодлогч (Hereditary Neuropathy with Liability to Pressure Palsies, HNPP) xoc суурын хэсэг дараалал нь делеци болсон байна. (Зураг-2,3)
- Статистик боловсруулалтын стандарт хазайлтын дундаж утгуудыг (Ct-value) тооцсон дүнгээр эмгэгийг нөхцөлдүүлэгч генийн дараалал агуулаагүй эрүүл хүнд диплойд (0.861.20) генотиптэй байна. Эмгэг өөрчлөлт бүхий сорьцонд Захын Миелин Ширхэгийн Уургыг кодлогч 1500 хос суурын урттай дараалал нь хоёрчлогдсон триплойд (1.40-1.93) генотиптэй харин удамшлын шалтгаантай Саа саажилтыг кодлогч хос суурын хэсэг дараалал нь делеци буюу гаплойд генотиптэй тодорхойлогдлоо.
Дүгнэлт
- Удамших хэв шинж, эмнэлзүйн үзлэг, ЭМГ-ийн шинжилгээ, молекул генетекийн шинжилгээгээр ШМТ болох тогтоогдлоо. Фридрихийн Атакси үгүйсгэгдсэн нь Зүүн Азийн орнуудад уг эмгэг ховор тохиолдог гэсэн бусад судлаачдийн үр дүнг бидний судалгаа давтаж байна.
- Захын Миелин Ширхэгийн Уургыг кодлогч ген, геномын хэсгийн хоёрчлогдсон байдал болон удамшлын шалтгаантай Саа саажилтыг кодлогч ген, геномын хэсгийн делеци өөрчлөлтүүд нь ШМТ-ийн 1А хэлбэрийн үндсэн шалтгаан болж байна.
Хэлцэмж
- Удамшлын мэдрэл булчингийн эмгэгүүд нь удамших хэв шинж, эмнэлзүйн шинж тэмдэгийн хувьд ижил төсөөтэй байдаг ч ЭМГ, орчин үеийн молекул генетикийн шинжилгээгээр тэдгээрийг өөрхоорондньялганоношлох.эмгэгийнхэлбэрийг нарийвчлан тодорхойлох бололцоог олгож байна. Гэвч ЭМГ-ийн шинжилгээ нь манай орны хувьд эмнэлзүйн практикт хараахан нэвтрээгүй байна.
- Удамшлын мэдрэл булчингийн эмгэгүүдийг ялган оношлож эцсийн шийдвэрлэх оношийг тодорхойлох нь цаашдын эмчилгээний тактикийг зөв оновчтой сонгох, эмгэгийн тархалт, бүтцийг тогтоох үндэс суурь болох төдийгүй урьдчилан сэргийлэх асуудлыг шийдвэрлэхэд ач холбогдолтой юм.
ШМТ-ийн хэлбэрүүд нь удамших хэв шинж, генийн мутац, эмнэлзүйн илрэлийн хувьд өөр хоорондоо ялгаатай байдаг. Өндер хөгжилтэй орнуудад анагаах ухааны орчин үеийн дэвшилтэт технологийг ашигласан ургийн хөгжлийн үед оношлох урьдчилан сэргийлэх ажилбарууд хийгдэж байна.
Үүнд:
А. Ихэсийн эс, эдийг шинжлэх
Б. Ургийн шингэн дэхь эс, эдийг шинжлэх
Удамшлын мэдрэл булчингийн эмгэг бүхий гэр бүлүүдэд оношлогоо, эмчилгээний асуудлыг хөндөж авч хэлэлцсэний дараагаар нөхөн үржихүй, гэр бүл төлөвлөлтийн талаарх удамзүйн зөвлөгөөг өгөх нь нэн чухал байна.
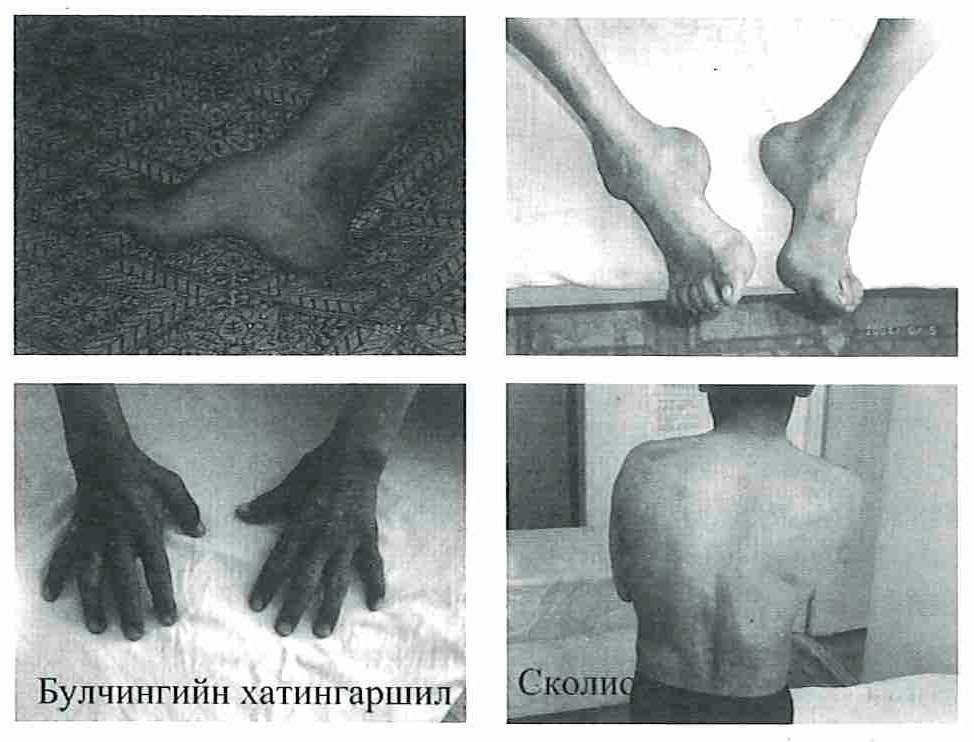
Зураг 1. Эмнэлзүйн шипж тэмдэгүүд Фридрих төст пшнхаи:
- Хонхор ул буюу тавхайн хонхор гүнзгийрэх
- Товгор (гүдгэр) гапуу
- Экапеизио - тавхайн угийн уенүүд уртсах, ялангуяа эрхий хуруу
- Флексио - тавхайн үзүүр үеээр богиносож хатингарших
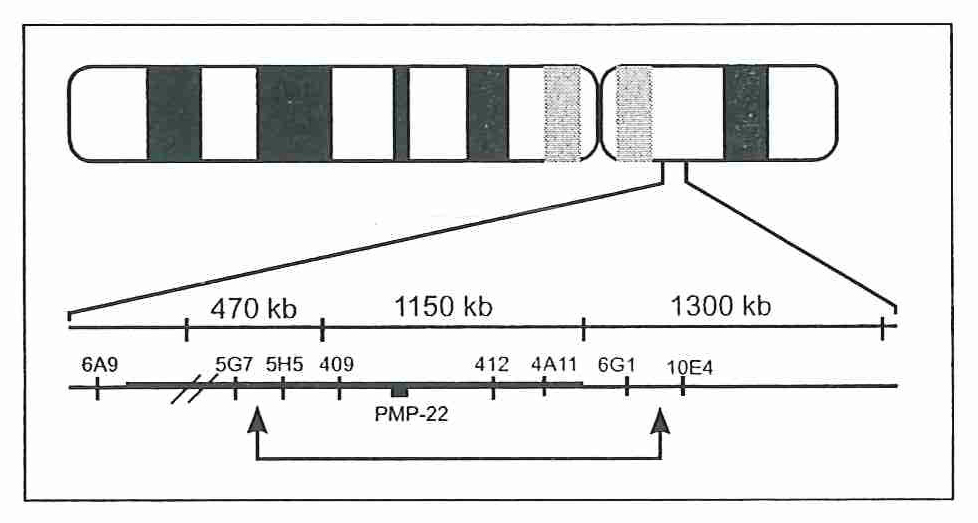
Зураг 2. Захьш Миелип Ширхэгийп Уургыг (Peripheral Myelin Protein 22, PMP22) кодлогч 1500 хос суурын урттай дараалал нь хоёрчлогдсон, Удамшлын Шалтгаантай Саа Саажилтыг кодлогч (Hereditary Neuropadiy with Liability to Pressure Palsies, HNPP) xoc суурын хэсэг дараалал иь делеци болсон байна.
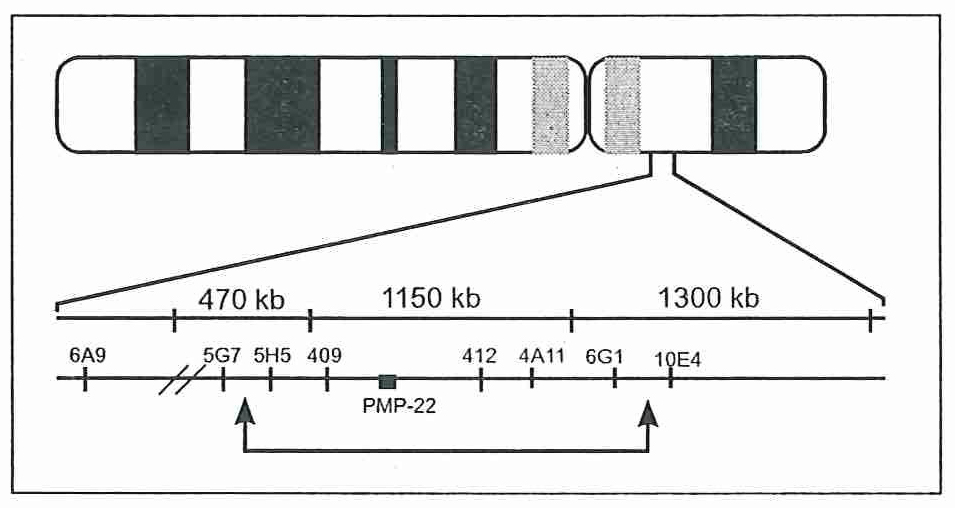
Зураг 3. Захын Миелин Ширхэгийн Уургыг (Peripheral Myelin Protein 22, PMP22) кодлогч 1500 xoc суурын урттай дараалал пь хоёрчлогдсон буюу диплойд геиотиптэй байхад гель электрофорез дээр нэмэлт зурвас тодорно
2. Баасанжав Д, бусад, УБ хотын хүн амын дунд мэдрэл удамшлын зарим өвчний тархалт, бүтэц, Монголын Анагаах Ухаан сэтгүүл, 2005, №4, х 11-13
3. Маркова Е.Д., Магжанов Р.В., Распространенность наследсвенных заболеваний нервной системы в различных популяциях, Жур. Невропат и псих. Им С.С. Корсакова, 1990, в9, с113-119
4. Skre Н., 1974, Genetic and clinical aspects of Charcot-Marie-Tooth\\\'s disease, Clin, genet., 6: 98-118
5. Emery A, E, H; Population frequencies of inherited neuromuscular diseases a world survey. // Neuromus. disord., 1991, 1: 19-29
6. Батчулуун Ш, Удамшлын мэдрэл булчингийн өвчний тархацын асуудал, «Анагаах Ухаан, эрүүлийг хамгаалах, нийгэм хангамжийн тулгамдсан асуудлууд» онол практикийн бага хурлын илтгэлийн хураангуй, 1990, х53-55
7. Martin В Detatycki, Robert Williamson, Susan M Forrest, Friedreich ataxia: An overview, Med Genet 2000; 37:1-8
8. Hum Genet, 2000, Analysis of Charcot-Marie-Tooth (CMT1A) and hereditary Neuropathy with liability to pressure palsies (HNPP), 107:494-498
9. Karen M, Krajewski MS, 2002, Charcot-Marie-Tooth disease, Gale Encyclopedia ofMedicine
10. Benjamin B. Roa, etal; 1993, Charcot-Marie-Tooth disease type 1A-Association with a spontaneous Point Mutation in the PMP22 Gene


